

PCV2 has emerged as one of the most devastating viral infections on swine farming, causing a relevant economic impact due to direct losses and control strategy expenses. PCV2 is the etiological agent of postweaning multisistemic wasting syndrome (PMWS), nowadays designated as PCV2-systemic disease (PCV2-SD, Figure 1). Epidemiological and experimental studies have evidenced that genetic diversity, characterized by global waves of emerging genotypes and the circulation of recombinant strains, may potentially affect the virulence of PVC2. Maybe that is the reason why the intra-specific classification of PCV2 has been historically controversial. In 2008, the EU consortium on Porcine Circovirus Diseases proposed a standardized nomenclature for PCV2 genotype definition based on pairwise sequence comparisons (www.pcvd.eu). The analyses performed to PCV2 complete and capsid (ORF2) nucleotide sequences defined two distance thresholds at 0.020 and 0.035, respectively. When the p-distance between two sequences was larger than those values it was assumed that the strains belonged to two different genotypes. The outcome of those analyses recognized four genotypes; namely PCV2a, PCV2b, PCV2c and PCV2d (also known as mutant PCV2b).


Figure 1. Three months pig with PCV2-SD. Note the marked spine, indicative of growth retardation, and body pallor (anemia).
Since 2008, a huge number of PCV2 sequences have been deposited in the GenBank (www.ncbi.nlm.nih.gov) and several new genotypes were proposed, though they were not always validated. A significant proportion of those sequences have a recombinant origin and in some cases they have been circulating with increasing prevalence in several Asian countries and USA. In a recent paper (Franzo et al, 2015), an international team of researchers re-analyzed the intra-specific taxonomy of PCV2 and the genotype definition to check their current validity in order to unify the nomenclature and avoid further misconceptions.
The obtained results indicated that, with the capsid and genome PCV2 sequences currently available, several assumptions of the methodology used to define the thresholds nearly a decade ago were violated. Specifically, the assumption of equal nucleotide substitution rates among the different PCV2 genotypes was not fulfilled. In addition, the definition of a single cut-off value to define PCV2 genotypes appeared complicated. More importantly, the thresholds proposed for the genotype definition would be undoubtedly meaningless (Figure 2). These evidences, coupled with the enormous amount of new sequences available, have important implications for PCV2 intra-specific classification. Hence, the thresholds applied since 2008 to define PCV2 genotypes are currently not applicable to all PCV2 strains and therefore the method should be re-considered.
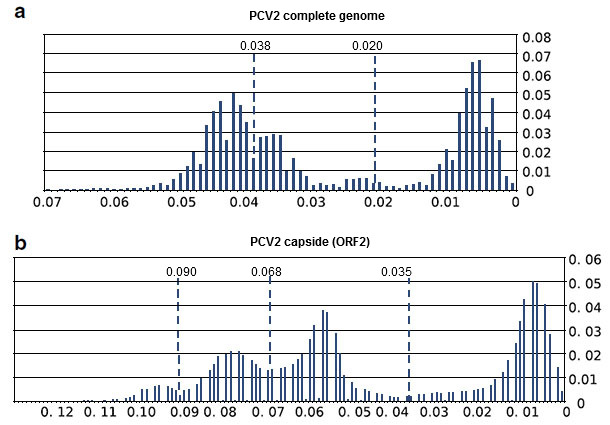
Figure 2. Threshold analyses of complete PCV2 genomes (a) and ORF2 sequences (b). The proportion of pairwise p-distances comprised within a 0.01 p-distance interval is reported. The proposed thresholds for the complete genome (0.020), the capsid (0.035), as well as the resulting thresholds of the re-analyses performed (0.038, 0.068 and 0.090) are also indicated.
Accordingly, an alternative method to genotype PCV2 strains in an unambiguous way is proposed. The suggested approach takes into account the presence of several recombinant strains that, as a matter of fact, belong to more than one genotype. Considering the high recombination frequency reported, a classification approach based on the ORF2 gene was preferred. Aiming at offering an unambiguous classification scheme, several reference sequences whose classification was clear for the phylogenetic methods were selected. This allows us to define four genotypes using both phylogenetic reconstruction and forty-seven genotype-specific marker nucleotide positions. Those marker positions consistently (>95%) characterize each genotype and can be used as a reference to assign a problem sequence to a certain genotype. The classification method proposed, apart from being robust, is very quick and easy to perform.
The taxonomical classification of PCV2 is still a real challenge. The results reported confirms and validates the variability of viral sequences and the high intra- and inter-genotype recombination frequency between PCV2 strains, highlighting the difficulty to pinpoint an unequivocal genotype definition for this virus. Moreover, the likelihood of emergence of new PCV2 genotypes in the future is very high, and new, robust genotyping methods might become a much needed tool.
The classification of PCV2 in genotypes is relevant on practical basis. So far, all available vaccines in the European and North-American market are based on PCV2a genotype, while the most prevalent ones are PCV2b and PCV2d ones. Although significant level of cross-protection among these three genotypes has been demonstrated, it would be interesting to assess if vaccine efficiency would be equivalent in front of all these different genotypes. Moreover, few phylogenetic studies published so far indicate that certain changes in aminoacids may occur in PCV2 sequences coming from vaccinated and non-vaccinated farms. In fact, it has been speculated the possibility that potential escape mutant viruses might be generated in the future due to such vaccination pressure. Therefore, proper genotype monitoring is key to assess vaccine efficacy.


